Rachel Aronoff recently raised an issue on the OFM software regarding fluorescence imaging. The thread has become far too general for the GitLab issue tracker, and so I’m making a copy here and moving the discussion.
Rachel Aronoff
We would like to be able to image ‘sybr-safe’ -stained cells with the openflexure scope.
we have gotten the gfp filter set from comar, and can get images with bright fluorescein, but so far have not been able to see the stained DNA in cells. Even when fixed cells are bright enough to see on our ‘diy trans-illuminator’ (with side-illumination), they do not show up with the openflexure system so far.
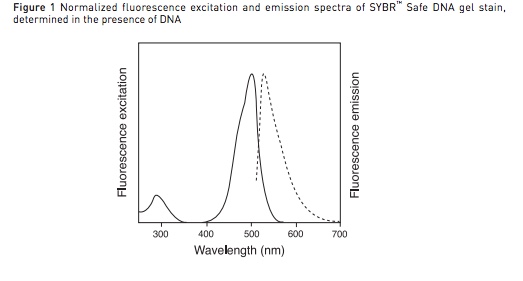
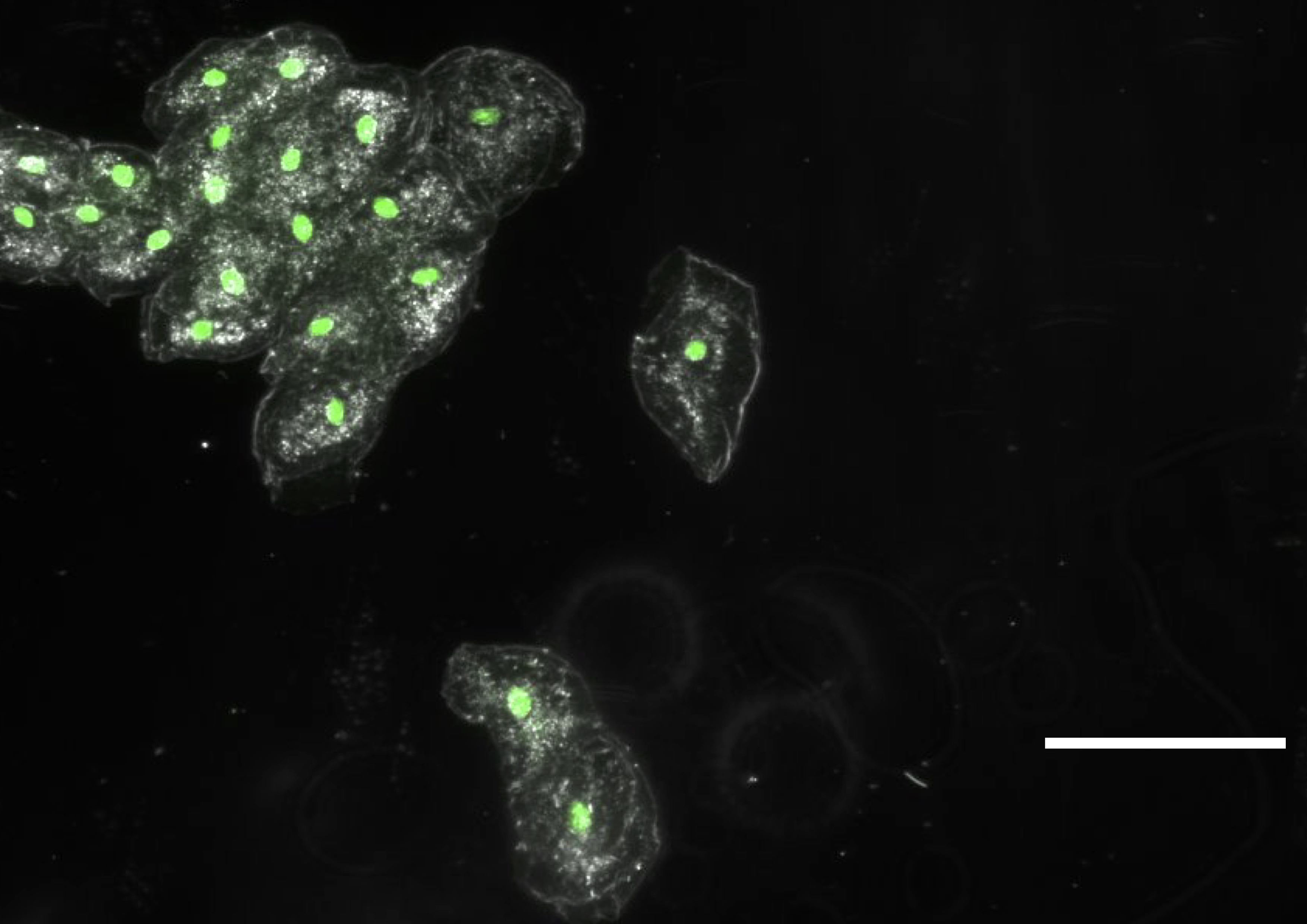
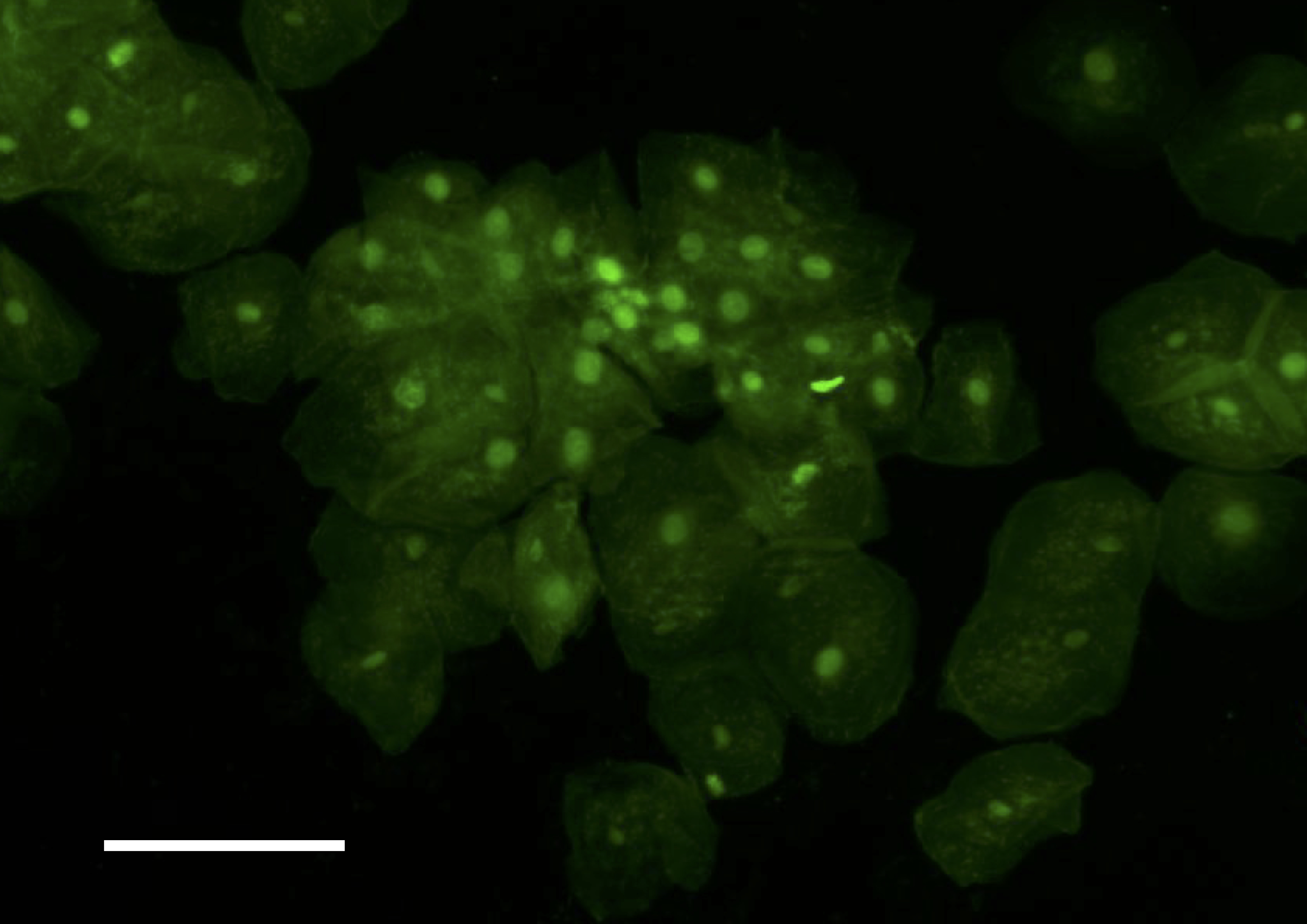
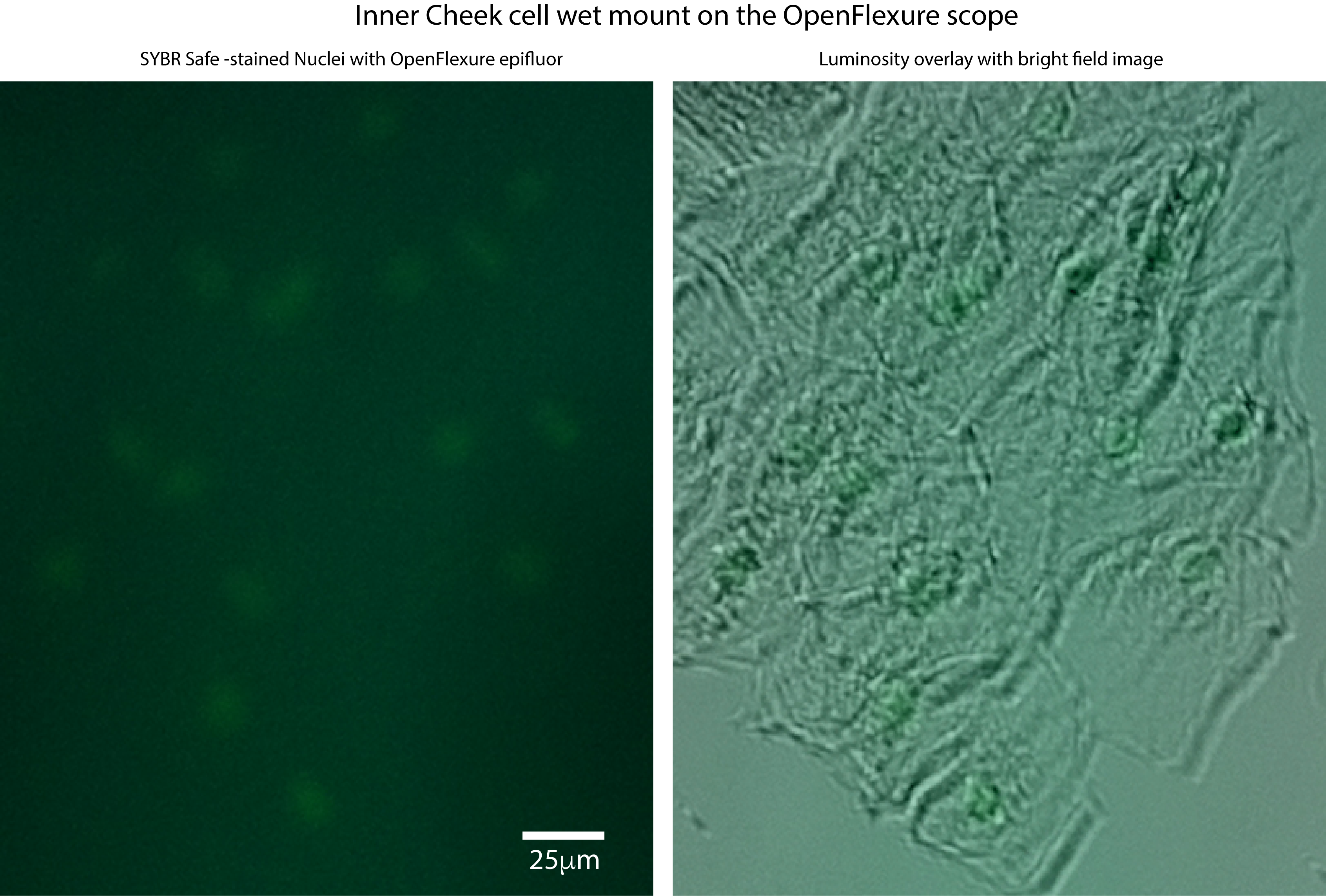
We are wondering if a sort of ‘binning’ could be tried, to average multiple images, thus reducing noise and increasing signal, in order to get the raspberry Pi camera to ‘see’ our stained cells. Can you get this to work, do you think? For your information, we have previously done such imaging with 500ms exposures on a standard lab epifluor setup and a 5x objective (when the bright field image is acquired with about 10ms exposures). I attach 4 pictures with the bright field and ‘epi’ images of cheek cells stained with sybr safe
, and of tissue, with fluorescein,
picture with 3 samples, the top, with the cheek cells stained with sybr safe, the left, tissue in water, and right the very bright tissue with fluorescein.
Thank you for your input and help.
p.s.(We also still worry that our 10X fluotar objective or the blue LED we are using for excitation might need to be changed…)
Rachel Aronoff @rachelaronoff · 1 week ago
btw, I added the arrows to show where nuclei should be bright in the epi image (for just a few of the cells)
Rachel Aronoff @rachelaronoff · 4 days ago
@jtc42 (Hi, Joel, Richard says you are the one to ask! ![]()
Do you have any ideas for us? As can be seen in the picture of the three slides through the ‘transilluminator’s’ orange filter, the fluorescein on the filter is super bright, but even that isn’t so impressive with the scope. Maybe we need to play more also with the camera calibration (any hints for that? - should it be done without a slide or with a non-fluor sample or with something fluor??), in addition to thinking about averaging images…? thx
Joel Collins @jtc42 · 4 days ago
The fact it’s so visibly bright in your photo but not so in the microscope images concerns me… Is there anything unusual in your filter setup that would explain that at all? Can you provide details on which filters you’re using, and how they’re set up in the microscope?
Regarding the camera calibration: While we’re still developing fluorescence imaging capabilities, we’re taking the approach of storing the raw bayer data in all our captures, and handling correction retroactively. This means we can test different calibration schemes without loss or re-taking data. For the time being, this is what I’d suggest.
We found that lowering the camera framerate, and using that to increase the exposure time helped a lot. Since the calibration is just a colour correction, I doubt you’d get any new information from that, only cleaner looking images. We had better results by increasing the exposure time massively. This will involve editing your ~/.openflexure/camera_settings.yaml file , as there is currently no option in the GUI to change framerate. It’s an easy change to make though. We got good results by setting it to 2fps, thus allowing a 500ms exposure. If you do this, make sure you have good vibration damping in place though!
Hopefully our work on fluorescence will continue in a week or so, and I’ll keep this thread updated if we make any developments.
Rachel Aronoff @rachelaronoff · 3 days ago
Thanks.
Will try the 2fps edit this week!
Wondering if I can also get the default back for the calibration (just in case? worried as we did many different calibs with different slides in place, and blank).
We have the standard Comar filters for GFP,
1 550nm dichroic mirror, 25x16mm (GFP) Mfr: Comar; Part No. 550 1Y 116
1 490nm excitation filter, 25x16mm (GFP) Mfr: Comar; Part No. 495 1K 116
1 500nm emission filter, 25x16mm (GFP) Mfr: Comar; Part No. 515 1B 116
but I am concerned that one (the dichroic esp) might be upside down - if that is possible…
We still have not gotten the motor boards together - I should have gotten the one from Valerian, I think, not just relied on our lab member (who has a new job and got very busy) - but getting the fluor is more impt than the panoramas for the moment!
![]()
Rachel Aronoff @rachelaronoff · 2 days ago
ok, we got the framerate changed to 2 (used nano for edit, and saved the original camera setting yaml…), and maybe sensitivity is a bit better, with exposure set at 5000 the fluorescein/tissue is very bright. but still the cells were not satisfactory. I will try tomorrow with fresh cells… (Also, there may be a bug: I noticed that when I set exposure to 6000 it goes to 5983 -ms?- and if I change the gain, then say ‘apply settings’ it goes back to 1.0) do you think I should try flipping the dichroic or test another blue led?? thx!
Joel Collins @jtc42 · 2 days ago
Hi Rachel, glad it helped! Do keep us updated.
The exposure not quite matching is normal, and unfortunately is not something we can prevent. When you set the exposure, it sets a target exposure which the camera hardware will try to match. Often it settles at a value that’s just a bit off.
I’m not sure if the gain should behave in the same way. I’ll look into this. It may be a bug in the client doing some nasty rounding
Rachel Aronoff @rachelaronoff · 1 day ago
thx? and is the dichroic I got uni-directional? should I try to flip it?
Rachel Aronoff @rachelaronoff · 1 day ago
tried the flip, and it is odd - I expected a big effect, but the cells are still not satisfactory, and the tissue with fluorescein still shows up just fine. with the big files (14mb each) I made overlays in photoshop, however, and maybe there are a couple of possible traces of potential nuclei, post flip, but it is hard to be convinced. Should I try to send these overlays with WeTransfer to you? Maybe trying another LED is the next step?? Any word from your end? Also, I did a series of changing exposures, but I would still like to know what the units are for those numbers, esp as changing the frame rate should have made for 500ms exposures… What is the actual difference in exposure when I go from about 10k to about 20k, without changing gains, for a specific question. (for the cells, I did the bright field with 10k and the ‘epi’ with 20k most of the time today, but for the fluorescein 10k for both was perfect…) I would also like to understand more what the calibration does, but guess I will also try more of this tomorrow. Can you look into the idea of ‘binning’ multiple images, to reduce noise and increase signal? Are there other people out there using the epi successfully for cells already, who I could contact? Thanks again for your help!
Joel Collins @jtc42 · 7 hours ago
with the big files (14mb each) I made overlays in photoshop
Files that big sound like you’re storing the raw bayer data in your captures. Please be aware that photoshop is unable to read this data. It can be extracted using Python but it’s really a picamera-specific block of data.
Should I try to send these overlays with WeTransfer to you?
You’re more than welcome to, but I’m not sure what I’ll be able to add really.
Maybe trying another LED is the next step??
If you have some brighter LEDs that will likely help. We’re going to be exploring using LEDs that kick out ~2W of power once our PhD student has started again.
Any word from your end?
We’ve all been away or busy (or ill) recently, and our PhD student working on the fluorescence has been on a 2 week break. He’s due to return next week, but will have a week of induction activities. Once he’s settled in, I think we’ll start working on the fluorescence again.
Also, I did a series of changing exposures, but I would still like to know what the units are for those numbers, esp as changing the frame rate should have made for 500ms exposures…
The value is returned as an integer representing a number of microseconds.
I would also like to understand more what the calibration does
We have a paper that should be released soon detailing what the calibration does. In the meantime, the file responsible for lens-shading functionality can be found here.
The routine is not super complicated, and the file has been heavily commented, so hopefully that’s useful.
Can you look into the idea of ‘binning’ multiple images, to reduce noise and increase signal?
This will almost certainly not appear in the microscope software, at least any time soon. We’re generally trying to keep data acquisition and data processing separate. What I might suggest we do is include a way to rapidly acquire a stack of images back-to-back without moving the stage. Then that stack of images can be overlaid in post.
Are there other people out there using the epi successfully for cells already, who I could contact?
As far as I’m aware, noone outside of the immediate project group is working on fluorescence imaging. @rwb27 might be able to confirm. At the moment, it’s heavily under development, so I consider this functionality very experimental.
Rachel Aronoff @rachelaronoff · 6 hours ago
well, PS did manage to open them for the overlays, although the exercise certainly did not clarify matters, and the final files were even more gigantic. Thank you for this further info, which I will look into further. I guess the question on the dichroic should also be addressed to Richard? (@rwb27)
Joel Collins @jtc42 · 5 hours ago
Sorry I should be clearer. The files contain a normal JPEG or TIFF (which PS can open and manipulate), but the majority of the data is additional raw bayer data that PS just discards. If the final files were also huge, that’s probably just down to whatever your PS export settings are set to.
RE the dichroic, Richard may have better advice than me, but if there’s absolutely no way to visually tell which side is which, then yeah maybe just trying both orientations is best. I’d be inclined to try and identify properly which way it should go though. The documentation may have useful information on that.
Richard Bowman ![]() @rwb27 · 3 hours ago
@rwb27 · 3 hours ago
I don’t think the dichroic should be particularly sensitive to which way round it goes. It’s probably best to have the coated side facing the microscope objective, but it won’t make a big difference. You can tell the coated side by holding it at 45 degrees and looking at the top edge of the optic - if you can see the edge clearly, you are probably not looking at it through the coated side of the dichroic.
Figuring out how to get the sensitivity up in fluorescence is definitely the hard part of all this. It’s good to see that you’ve got it working with GFP - but it’s odd that it looks so bright in your side-illuminated set-up and not in the microscope. If you side-illuminate the sample in the microscope, what do you see? I have had issues in the past with leakage of excitation light into the microscope doing that, so it’s not 100% trustworthy, but it might be worth a try.
Rachel Aronoff @rachelaronoff · just now
it worked not with GFP, but with the chemical fluorescein. that is why it is so very bright. how to get the sybr stained cell nuclei to show up is really the key thing that we need, for any of this to be worthwhile… yesterday’s fresh wet cells were no where near as bright as the ones shown above (dry and with tons of sybr)… will try adding a side illumination. hmmm. (and double check the dichroic coating) so you concur that the calibration is not so helpful?? I wonder if a previous calibration done is messing up the acquisition now, and am sad that averaging images does not seem readily feasible…
Joel Collins @jtc42 · just now
Hi all,
I think this thread would be better suited to the GOSH forum at this point. It’s become far too general to using the microscope, where this is an issue tracker for the server-side part of the microscope software specifically.
I’m going to create a GOSH thread on this, and once that’s online, I’ll close and lock this issue so we can move general discussion there.
and am sad that averaging images does not seem readily feasible…
If this is something you’re particularly interested in, could you please raise a specific issue about this? That way, we can assign the feature to a release and it might get included at some point. It’s important to state that because of the way our camera streams work, it’s not trivial to add, so a separate issue would be ideal.


 )
)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}